
X-Ray crystallography is a technically sophisticated but conceptually simple-minded method with the great advantage that, to a first approximation, its results are independent of whatever preconceptions we bring to the task. This was very fortunate in the case of proteins, because it is unlikely that we could ever have successfully made the jump to such elegant and complex structures as those shown in Figs. 1 and 2 if we had been obliged to rely on more logical and indirect methods. For small inorganic and organic molecules indirect inference had succeeded magnificently, so that X-ray crystallography provided no startling revelations but only a prettier and more accurate picture of what was already known. However, even after knowing what the answer should look like for proteins, 20 years of effort has failed to derive three-dimensional protein structures from spectroscopic and chemical data or from theoretical calculations.


FIG. 1. Schematic drawing of the polypeptide backbone of ribonuclease S (bovine pancreatic ribonuclease A cleaved by subtilisin between residues 20 and 21). Spiral ribbons represent α-helices and arrows represent strands of β sheet. The S peptide (residues 1-20) runs down across the back of the structure.
[Even after more than 40 years, we have made significant progress but still have certainly not solved the problem of predicting structure from sequence. The biggest change has been homology modeling: if a structure is known for a related sequence, which is now increasingly likely, then an approximate structure can be built which is useful for many purposes. De novo prediction is now sometimes quite close, but is certainly not reliable or routine. The prediction effort has been enhanced, and can be followed, through the Critical Assessment of Structure Prediction (CASP) competition (e.g., () .]
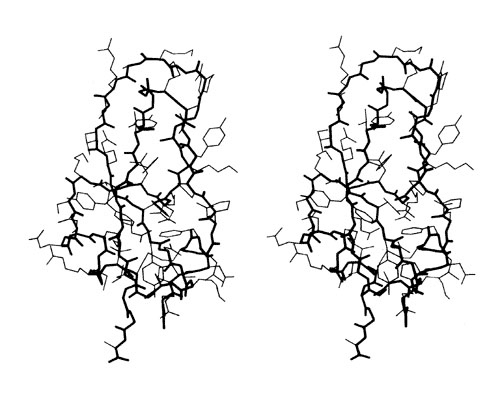
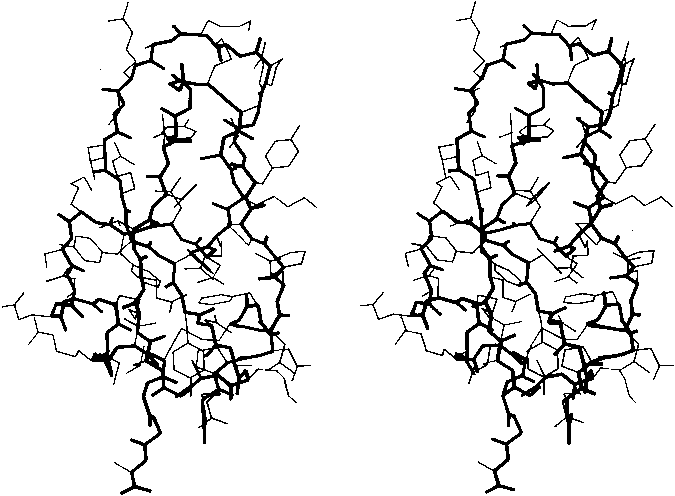
FIG. 2. Stereo drawing of all nonhydrogen atoms of basic pancreatic trypsin inhibitor. The main chain is shown with heavy lines and side chains with thin lines.
Before the first X-ray results, protein structure was visualized in terms of analogies based on chemistry and mathematics. The models proposed were relatively simple and extremely regular, such as geometrical lattice cages (), repeating zigzags (), and uniform arrays of parallel rods (). In light of these very reasonable expectations, the low-resolution X-ray structure of myoglobin () came as a considerable shock. Kendrew, in describing the low-resolution model (see Fig. 3), says "Perhaps the most remarkable features of the molecule are its complexity and its lack of symmetry. The arrangement seems to be almost totally lacking in the kind of regularities which one instinctively anticipates." Perutz was even more outspoken about his initial disappointment: "Could the search for ultimate truth really have revealed so hideous and visceral-looking an object?" ().

FIG 3. Electron density contours of sperm whale myoglobin at 6 Å resolution.
In the last 20 years we have learned to appreciate the aesthetic merits of protein structure, but it remains true that the most apt metaphors are biological ones. Low-resolution helical structures are indeed "visceral," and high-resolution electron-density maps (for instance, see Fig. 13) are like intricate, branched coral, intertwined but never touching. β sheets do not show a stiff repetitious regularity but flow in graceful, twisting curves, and even the α-helix is regular more in the manner of a flower stem, whose branching nodes show the influences of environment, developmental history, and the evolution of each separate part to match its own idiosyncratic function.
The vast accumulation of information about protein structures provides a fresh opportunity to do descriptive natural history, as though we had been presented with the tropical jungles of a totally new planet. It is in the spirit of this new natural history that we will attempt to investigate the anatomy and taxonomy of protein structures.