
Many protein molecules are composed of more than one subunit, where each subunit is a separate polypeptide chain and can form a stable folded structure by itself. The amino acid sequences can either be identical for each subunit (as in tobacco mosaic virus protein), or similar (as in the α and β chains of hemoglobin), or completely different (as in aspartate transcarbamylase). The assembly of many identical subunits provides a very efficient way of constructing large structures such as virus coats. Often a multisubunit molecule is more smoothly globular than its component subunits are, as for instance in the insulin hexamer shown in Fig. 61.
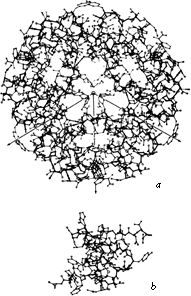
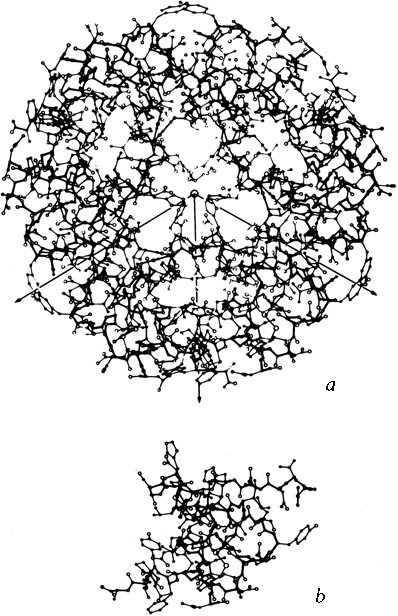
FIG. 61. (a) The insulin hexamer; (b) the insulin monomer. From Blundell et al. (), with permission.
The surfaces that form subunit-subunit contacts are very much like parts of a protein interior: detailed fit of generally hydrophobic side chains, occasional charge pairing, and both side chain and backbone hydrogen bonds. Twofold symmetry is the most common relationship between subunits. The 2-fold is often exact and can be part of the actual crystallographic symmetry, as for the prealbumin dimer in Fig. 62. However, in many cases (e.g., ; ) individual side chains very close to the approximate 2-fold axis must take up nonequivalent positions in order to avoid overlapping (see Fig. 63). Conformational nonequivalence can extend further away from the axis and produce such effects as different binding constants for ligands (e.g., ). Tetrahedral 222 symmetry is also common, either with only one or with all three 2-folds exact (e.g., ).
A rather common feature of subunit contacts is β sheet hydrogen bonding between strands in opposite subunits. Theoretically the relationship could be a pure translation or a 2-fold screw axis with a one-residue translation (for a pair of parallel strands), but all the known cases of inter-subunit β sheet bonding turn out to be between equivalent strands related by a local 2-fold axis. For hydrogen-bond formation, the 2-fold must be perpendicular to the β sheet, requiring the two equivalent strands to be antiparallel. Those may be the only two β strands (as in insulin, Fig. 63), or they may be part of antiparallel β sheets (as in prealbumin, Fig. 62), or the rest of the sheets may be parallel (as in alcohol dehydrogenase domain 1).
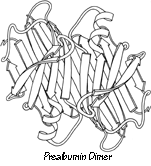
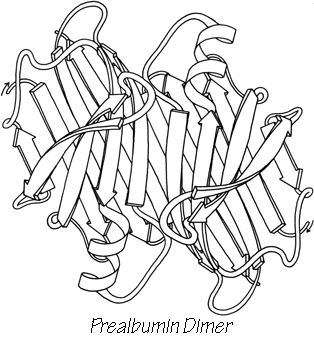
FIG. 62. A schematic drawing of the backbone of the prealbumin dimer, viewed down the 2-fold axis. Arrows represent β strands. Two of these dimers combine back-to-back to form the tetramer molecule.
Similar subunit structures can assemble in quite different ways. The Greek key β barrel of Cu,Zn superoxide dismutase assembles back-to-back across a tight, hydrophobic side chain contact, while the Greek key β barrel of prealbumin joins by continuing the β sheet bonding side-by-side. Even for proteins known to be closely related the subunits may associate in nonhomologous ways, such as the monomers versus tetramers in various hemoglobins, or the atypical contact between chains in the Bence-Jones protein Rhe (). The homologous domains in the immunoglobulin chains associate in three quite different types of pairwise contact: the usual "back-to-back" barrel contact of VL and VH (shown in Fig. 101); CL and CH1 barrels contact "front-to-front"; and pairs of CH2 domains are quite widely separated by carbohydrate (). Of course, the great majority of homologous proteins have homologous subunit contacts, but it seems that even a quite drastic change in subunit contacts is easier to accommodate than major internal rearrangement.
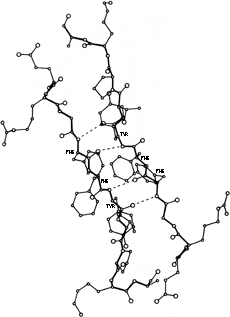
FIG. 63. Departures from local 2-fold symmetry, especially of side chain positions, in the β strand dimer interaction of insulin. From Blundell et al. (), with permission.
Twofold contacts are self-homologous—formed by equivalent surfaces from each of the participating subunits, while the occasional 3-fold (e.g., bacteriochlorophyll protein), 4-fold (hemerythrin), or 17-fold (tobacco mosaic virus) contact is heterologous—formed by joining two different surfaces. An especially interesting type of heterologous contact has been found in hexokinase (). It was previously assumed that self-associating arrays with a definite number of subunits had to be related by a closed, point-group symmetry operation in order to avoid producing infinite aggregation (e.g., ). Hexokinase, however, has a 156° rotation plus a 13.8Å translation between subunits, which demonstrated clearly that screw axes are also permissible as long as addition of a third subunit by the same screw operation is blocked by overlap with the first subunit. Such a screw-axis relationship can easily produce very marked nonequivalence between chemically identical subunits. [Now that structures are routinely done for large complexes, rotational symmetry is seen quite often, Three-folds (e.g. membrane porins: 2OMF), four-folds (e.g. K+chain: 1BL8), and five-folds (e.g. choleratoxin: 3CHB) are common, and many others occur such as the seven-folds of Gro-EL (1DER), the nine-folds of the light-harvesting complex (1LH2), and the II-folds of TRAP protein (1C9S). In addition to the icosahedral viruses, many other complexes have cubic symmetries, such as ferritin (1AEW) or glutamine synthetase (1FS2).]
Subunit contacts need to be relatively extensive and stable if they are to ensure subunit association in the absence of a covalent link. However, in some cases a subunit contact can shift back and forth between two different stable positions, as has been demonstrated for oxy- versus deoxyhemoglobin (). Allosteric control can then be exerted by any factors which either affect the local conformation or bind between the subunits. A less elegant but even more extreme example is lamprey hemoglobin, which dissociates altogether in the oxy form ().
Subunit motion between two positions is also critical to the assembly of tobacco mosaic virus. In the partially assembled "disks," having two stacked layers of 17 subunits each, the layers are wedged apart toward their inner radius. During assembly of the viral helix, RNA binds between the layers, which then clamp tightly together with 16(1/3) subunits per turn (; ).
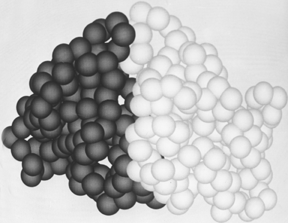
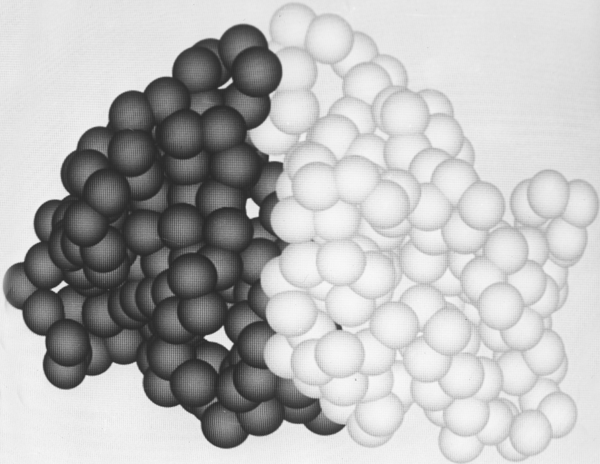
FIG. 64. The tightly associated domains (one shown light and the other dark) of elastase. Figures 64 through 66 use a space-filling representation with a sphere around each α-carbon position; they were photographed from Richard Feldmann's molecular graphics display at the National Institutes of Health.
Within a single subunit, contiguous portions of the polypeptide chain frequently fold into compact, local, semi-independent units called domains. The separateness of two domains within a subunit varies all the way from independent globular domains joined only by a flexible length of polypeptide chain to domains with extremely tight and extensive contact and a smoothly spherical outside surface for the entire subunit (such as in Fig. 64). An intermediate level of domain separateness is common in the known structures, with an elongated overall subunit shape and a definite neck or cleft between the domains, such as phosphoglycerate kinase shown in Fig. 65.
Another feature frequently seen in both domain and subunit contacts is an "arm" at one end of the chain which crosses over to "embrace" the opposite domain or subunit. Figure 66 shows such "arms" on the domains of papain. "Arms" that cross between domains or subunits almost invariably lie at the surface, but one unusual case in influenza virus hemagglutinin has a piece from a different domain forming the central strand of a five-stranded β sheet (see Fig. 83, where the alien strand is shown by the dotted lines).


FIG. 65. The "dumbbell" domain organization of phosphoglycerate kinase, with a relatively narrow neck between two well-separated domains.
The paucity of examples of flexibly hinged domains is almost certainly due to the difficulties of crystallizing such structures. In the immunoglobulins it has long been known from electron microscopic and hydrodynamic evidence that the hinge between the Fab and Fc regions is very flexible. The intact Dob immunoglobulin whose structure has been determined () has a substantial deletion in the hinge region which presumably limits its flexibility greatly. Intact immunoglobulins without such a deletion are notoriously difficult to crystallize, and the two cases in which crystallization has succeeded both turned out to have ordered Fab regions and invisible, disordered Fc portions (; Edmundson, 1980). A study of diffracted X-ray intensity as a function of resolution has shown that the Fc disorder is probably a static, statistical disorder among at least four multiple conformations (). [More recently, two more intact immunoglobulin structures have been solved, one hinged into a Y shape (1IGY) and the other a T shape (1IGT). ] This motion of flexibly-linked domains is illustrated in Fig. 66x and the associated kinemage ?? BnT c15,k2.
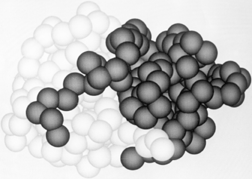
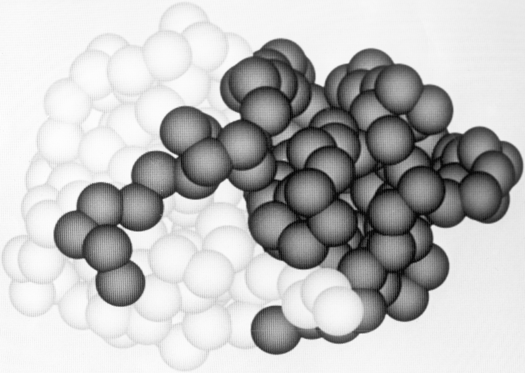
FIG. 66. The domains of papain, which wrap "arms" around each other.
At the other extreme, with very tightly associated domains, it is rather difficult to make the decision as to how many domains should be said to be present. In naming domains for the present study we have made use wherever possible of experimental evidence about either hinge motions of domains or about their folding or stability as isolated units. For example, rigid-body hinging has been documented for hexokinase (), phosphoglycerate kinase (), tomato bushy stunt virus (), and between immunoglobulin VL and CL domains (; ). On the other hand, the known movements on substrate binding in carboxypeptidase (), adenylate kinase (Sachsenheimer and Schulz, 1977), and phosphorylase () involve only surface movement of loops. For elastase () it is known that after proteolytic cleavage the domains can fold up into stable isolated units. But on the other hand, none of the large fragments of staphylococcal nuclease () or of cytochrome c () can fold up independently. For the majority of proteins, where such experimental evidence is not available, the decision about domains was made on the basis of analogy: whether the whole subunit or its parts more closely resembled other single-domain proteins. It is possible that some of the larger domains listed here will turn out to have genuinely independent smaller parts; domain divisions have been claimed within subtilisin (; ) and the first domain of phosphorylase (). Several domains of more than 300 residues are now known, however, which cannot plausibly be subdivided [e.g., triosephosphate isomerase, carboxypeptidase, bacteriochlorophyll protein]. Since large domains are clearly possible, we have been conservative in assigning divisions. Only three of the domains consist of two long, noncontiguous segments (e.g., pyruvate kinase d1); most are a single piece of chain.
The above definition of domains, in which they are thought of as potentially independent, stable folding units analogous to subunits, is only one of three rather separate concepts of domains in current thinking about the subject. The initial recognition of domain divisions as a general feature in the three-dimensional structures () was in terms of locally compact globular regions also contiguous in the sequence. This idea has recently been elaborated and computerized in terms of maximum long-range contacts between segments with only short-range internal contacts (), binary divisions of the sequence that maximally lie on opposite sides of a single cutting plane (), or binary sequence divisions that minimize the sum of the separate surface areas (). Each of these algorithms agrees in many cases with "intuitive" or "subjective" division into domains, but one of the more striking results of this kind of analysis is that it always produces a hierarchy or tree of substructures, usually containing two or three levels between the individual secondary-structure elements and the entire subunit. Domains as usually conceived represent the upper levels of such a hierarchy, while the lower levels may very well represent intermediates in the folding process. One lesson from these studies, however, is that something else is involved in the intuitive concept of a domain besides these purely geometrical criteria of relative compactness. In the current study we have provided that additional criterion by requiring analogy to some structure known to possess stability in isolation.
The third major concept of domains is basically genetic, following quite naturally from the earliest idea of domains: the homologous, internally disulfide-linked regions in the immunoglobulin sequence (). Rossmann () has proposed that the similar nucleotide-binding domains in various dehydrogenases represent genetic segments that have been transferred and combined with differing catalytic domains to produce functionally distinct but partially related enzymes. Recently the discovery of exon (translated and expressed) DNA sequences separated by intron sequences which are clipped out of the mRNA before translation into protein has raised the intriguing possibility that separate exons correspond to structural and/or functional domains recognizable in the proteins and basic to their evolutionary history. In immunoglobulins the exons correspond quite exactly with structural domain divisions, with the hinge region as a separate exon (). However, in hemoglobin the introns occur not only inside what is usually considered a single domain, but even in the middle of individual helices, although it has been found that the isolated central exon peptide can bind heme (). At the current level of knowledge it is unclear whether exons will provide a clarification of the basis of structural domains, although they are clearly a fundamental breakthrough in our understanding of the evolutionary processes involved.
Domains have proved so fruitful in explaining both the structure and the function of proteins that the concept is certain to survive in one form or another. At some time in the near future it will presumably acquire full scientific respectability with a verifiable definition in terms of either folding units or genetic units or perhaps both. [In practice, there are now two accepted standards at opposite ends of the domain problem. Genomics and bioinformatics now allow routine, reliable identification of functional domains that recur in many different sequence contexts, often as repeats but occasionally as isolated subunits; a few examples are zinc fingers, pleckstrin homology domains, and fibronact in type 3 domains. At the other extreme, of difficult and perhaps unique cases in individual structures, the de facto standard is the assignments made by Alexci Murzin for the SCOP database (http://scop.mrc-lmb.ac.uk/scop).]
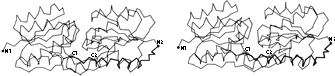
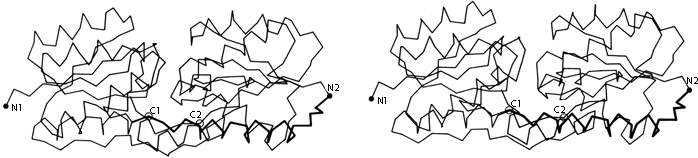
FIG 67. Stereo α-carbon drawing of the two domains of arabinose-binding protein (viewed perpendicular to the approximate 2-fold axis between domains), with the stretch of chain shown dark which joins the end of the first domain to the beginning of the second one.
The domains (as we have defined them) within a subunit very often resemble each other (as discussed in Section IV,B), and those similar domains are frequently related by an approximate 2-fold axis. The 2-fold relationship often occurs even in cases in which it involves considerable inconvenience because the start and end of the chain are on opposite sides of the basic domain structure, so that a long additional loop is required to connect the end of the first domain to the beginning of the second one (see for instance Fig. 67). An even more interesting instance of the pervasiveness of 2-fold domain contacts is in the serine proteases, where the relationship between genetically equivalent portions of the chain is in fact a pure translation. However, the initially heterologous contact seems to have evolved toward 2-fold similarity of the contact surfaces, so that now these proteins give the appearance of having a 2-fold relationship around the midpoint of the sequence (see Fig. 68). This convergent evolutionary process was able to produce an apparent sequence inversion because the topology of the serine protease domains (+1, +1, +3, -1, -1, or -1, -1, -3, +1, +1) is invariant to reversal of N- to C-terminal direction. The kinds of cases illustrated in Figs. 67 and 68 suggest that 2-fold contacts are inherently easier to design well than unmatched, heterologous contacts.
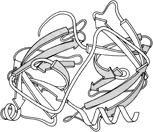
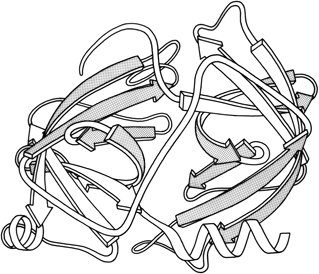
FIG. 68. Schematic backbone drawing of the elastase molecule, showing the similar β barrel structures of the two domains. The outside surfaces of the β barrels are stippled.
The types of contacts between domains are in general very similar to those found between subunits, but there are a few characteristic differences. Continuation of β sheet hydrogen-bonding, which is very common between subunits, is almost unknown between domains (which typically contact through side chains rather than main chain). β barrel domains (see Section III,D) most often associate side-by-side, usually with their barrel axes at a 50 to 90° angle to each other (as in Fig. 68). Doubly wound parallel β sheet domains (see Section III,C) most often associate with the β strands pointing toward each other (as in Fig. 68).
Domains as well as subunits can serve either as moving parts for the functioning protein or as modular bricks to aid in efficient assembly. Undoubtedly the existence of separate domains is important in simplifying the folding process into separable, smaller steps, especially for very large proteins. The commonest domain size is between 100 and 200 residues, but it now appears that there is no strict and definite upper limit on practical folding size: domain sizes vary by an order of magnitude, from 41 residues up to more than 400. The range of domain sizes is somewhat different for each of the major structural categories (see Sections III,B through III,E for definitions): generally less than 100 residues for small disulfide-rich or metal-rich proteins, 80 to 150 for antiparallel α, 100 to 200 for antiparallel β, and 120 to 400 for parallel α/β. The lower limit for each category presumably reflects the smallest stable structure that can be made using that general design pattern. The upper limit may, among other things, reflect the largest domain for each structure type that can fold up efficiently as a single unit.
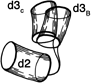
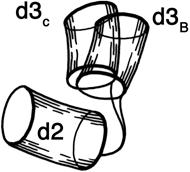
FIG. 69. The two different positions of the hinge between domains 2 and 3 of tomato bushy stunt virus protein. Each domain is represented by a cylinder, with domain 2 as the reference and domain 3 shown in the relative positions it takes in type B subunits and in type C subunits.
The other important function of domains is to provide motion. Completely flexible hinging would be impossible between subunits because they would simply fall apart, but it can be done between covalently linked domains. More limited flexibility between domains is often crucial to substrate binding, allosteric control, and assembly of large structures. In hexokinase the two domains hinge together on binding of glucose, enclosing it almost completely () [see kinemage ??.] . Not only does this mechanism provide access to a very tight and hydrophobic specificity site, it has also been hypothesized as necessary in order to discriminate against using water as a substrate and acting as a counterproductive ATPase. It should be remembered, however, that domain motion is not the only way to solve the problem: what hexokinase and alcohol dehydrogenase accomplish by domain hinging, adenylate kinase and lactate dehydrogenase accomplish by large movements of surface loops. [Domain hinge motion has remained extremely common, especially for enzymes, many of which show the overall pattern of two lobes with the active site in a cleft between them.]
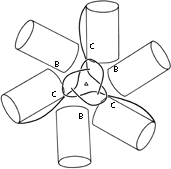
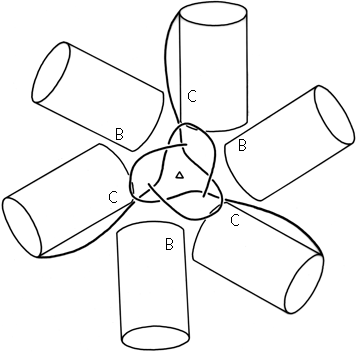
FIG. 70. Domains 2 (cylinders) and N-terminal tails of B- and C-type subunits around the quasi-6-fold axis in tomato bushy stunt virus. The association of the three C-subunit tails around the quasi-6-fold forms "domain 1" (see Fig. 84).
In tomato bushy stunt virus protein, domain hinging helps to solve the problem of packing 180 identical protein subunits quasi-equivalently into the 60-fold icosahedral symmetry of the virus shell (). Around the exact 2-fold and the quasi-2-fold axes of the icosahedron, contact must be made between chemically identical subunits which come together at rather different angles around the two kinds of 2-fold axes. Identical contacts are made in both cases between pairs of protruding d3 domains, but the angle between d2 and d3 domains can change by about 20° (see Fig. 69) so that each type of d2 domain is placed in the correct orientation to interact around the 5-fold or the quasi-6-fold axes. Pairs of d2 shell domains form two different types of contacts: in one type the two surfaces are tightly packed and the N-terminal arms are disordered, while in the other type of contact the N-terminal arms of "C" subunits fill a wedge-shaped opening between the contact surfaces and then wind around the 3-fold axis (see Figs. 70 and 84).