
In a very real sense, the structure of the closely bound water molecules around a protein are a part of the protein structure: they determine conformation of the exposed side chains, stabilize the ends of secondary structures, and occupy positions at active sites where they influence substrate binding and sometimes catalysis. The properties of the bulk water are critical in stabilizing the folded native form of proteins (e.g., ), but it is only the bound water that we will consider to be an actual part of, rather than an influence on, the protein structure.
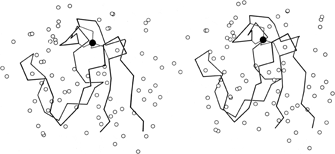
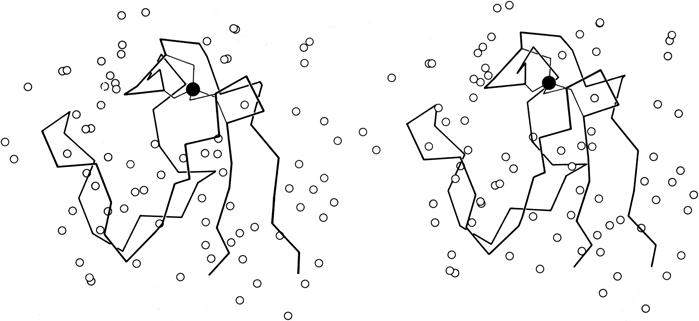
FIG. 58. Stereo drawing of the rubredoxin backbone with the iron (filled circle) and its cysteine sulfur ligands and all the water molecules (open circles) identified during refinement of the structure at 1.2Å resolution. Adapted from Watenpaugh et al. (), Fig. 11, with permission.
In high-resolution X-ray structures of proteins it is usual for a small number of solvent molecules to appear fairly clearly as peaks in the electron density map (see Fig. 13). Now that various refinement techniques are being applied to many protein structures, determination of water positions is usually a part of the process. In only a few cases, such as the study of rubredoxin in Watenpaugh et al. () and the study of actinidin in Baker (), has a real attempt been made to locate all of the fairly tightly bound waters and to eliminate spurious peaks. Figure 58 shows the waters around rubredoxin. Occupancies as well as positions are refined so that partially ordered as well as tightly bound water can be located. It is in fact only relatively few waters for which the occupancy approaches 1 (23 of the 130 waters located in rubredoxin had occupancies ≥ 0.9). [Identification and refinement of waters is now quite standard, and except at low resolution they would usually be assigned in numbers on the order of one per amino acid residue. Note, however, that occupancies and B-factors cannot really be cleanly separated for waters; in some structures these days only B is refined and all waters are left at 1.0 occupancy, but this does not of course mean that they are always present.]
Another recent study which provides less direct, but also very detailed, information about the water around a protein is the Monte Carlo calculations performed by Hagler and Moult () for egg lysozyme. From random starting positions they obtain a very long series of possible sets of water positions for which the statistical properties must obey all the constraints of the energy functions used. Contour maps can be plotted giving the overall frequency of water location at each point, and they match the refined X-ray electron density contours quite well. Also, individual sets of positions at single cycles in the simulation can be examined. The energies of water molecules in various types of locations can be determined for the overall simulation and can be compared with the energy distribution for the bulk water.
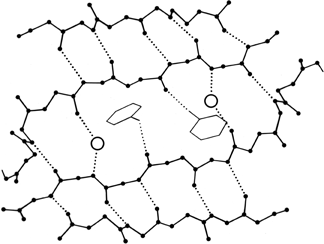
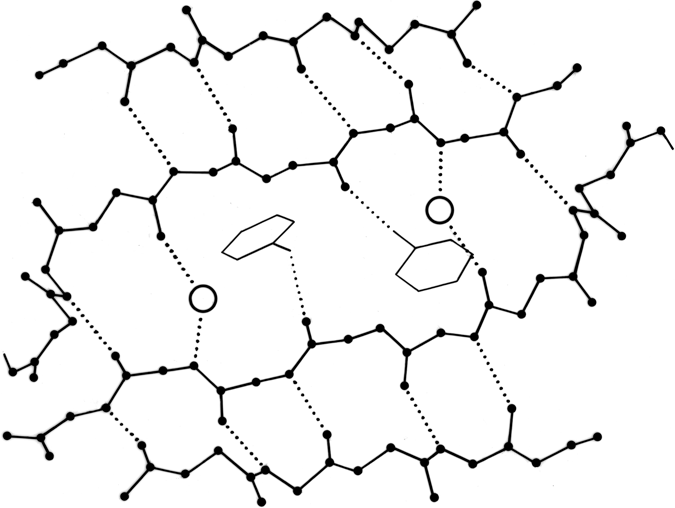
FIG. 59. Water molecules (open circles) in prealbumin, bridging between main chain groups that are too far apart to continue β-type hydrogen-bonding between strands. A hydrogen bond to a tyrosine side chain is also shown.
The detailed study of water structure around proteins is only just beginning, but a number of conclusions can be drawn from the crystallographic and theoretical work that has already been done. Isolated water molecules occur trapped inside protein interiors, where they can fill defects in the side chain packing and usually make some hydrogen bonds to protein atoms. Their energies are rather high, but it is much better to have a water than an empty hole in those locations. The number of such internal waters varies very widely from one protein to another. Both for internal and for surface waters, it is very common that they bond to the first free backbone NH or CO groups at the ends of pieces of secondary structure; for β strands it is common that the last H-bond opens up wider, with a water bridging in between (see Fig. 59).
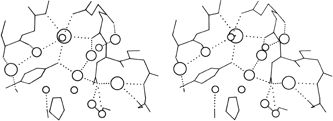
FIG. 60. A stereo view of one of the hydrogen-bonded networks of water molecules at the surface of the rubredoxin molecule [adapted from Watenpaugh et al. (), Fig. 7, with permission]. The size of the waters is proportional to their occupancy factors, so that the most well-ordered waters are shown largest.
The most ordered surface waters are those around charged side chains or in surface crevices. Occasionally those crevices can be very deep, such as the active site pocket in carbonic anhydrase, which extends about 15 Å in from the surface, with a network of water molecules (). The well-ordered waters at the protein surface are usually part of an approximately tetrahedral (but sometimes planar trigonal) network of hydrogen bonds to the protein and to other waters. An example from rubredoxin is shown in Fig. 60.
Both crystallographically and also from vapor-pressure measurements of solvent stabilization () it appears that water hydrogen bonds more frequently and more strongly to peptide CO groups than NH groups. In rubredoxin, only 24% of the available backbone NH groups are bonded to water and 70% to other protein atoms, while for the CO groups 43% bond to waters, 41% to protein atoms, and another 8% to both ().
Many of the tightly bound waters have energies substantially lower than the bulk water (). All studies have found that most of the bound waters, and all of the highly ordered ones, are in the first coordination layer, but that they do not by any means cover the whole protein surface. A substantial number of partially ordered waters are found in the second coordination layer (where they hydrogen-bond to the protein only through first-layer water) and essentially none any further out than that, even where there are suitably sized channels between neighboring protein molecules. The degree of motion seen for individual water molecules also increases dramatically as a function of their distance out from the protein. [Waters, and exposed side chains as well, are more ordered at the cryogenic temperatures now standardly used for data collection, and they are also more ordered in crystal contacts. The molecular contacts in a crystal are usually rather weak, with relatively sparse contact between protein atoms but many contacts through single ordered waters. Such contacts can even have hydrophobic atoms on both sides with rings or clusters of waters in between, as seen in the very high-resolution crambin structure (Teeter).]