
The α-helix is the classic element of protein structure. A single α-helix can order as many as 35 residues whereas the longest β strands include only about 15 residues, and one helix can have more influence on the stability and organization of a protein than any other individual structure element. α-helices have had an immense influence on our understanding of protein structure because their regularity makes them the only feature readily amenable to theoretical analysis.
FIG. 11. Drawing of a typical α-helix, residues 40-51 of the carp muscle calcium binding protein. The helical hydrogen bonds are shown as dotted lines and the main chain bonds are solid. The arrow represents the right-handed helical path of the backbone. The direction of view is from the solvent, so that the side groups on the front side of the helix are predominantly hydrophilic and those in the back are predominantly hydrophobic.
The α-helix was first described by Pauling in 1951 () as a structure predicted to be stable and favorable on the basis of the accurate geometrical parameters he had recently derived for the peptide unit from small-molecule crystal structures. This provided the solution to the long-standing problem of explaining the strength and elasticity of the α-keratin structure and accounting for the appearance of its X-ray fiber diffraction pattern. Helices had frequently been proposed before as the α structure, but none of them could adequately match the diffraction pattern because they had been limited by the implicit assumption that a regular helix would necessarily have an integral number of amino acid residues per turn. In fact, as Pauling first realized, the α-helix has 3.6 residues per turn, with a hydrogen bond between the CO of residue n and the NH of residue n + 4 (see Fig. 11). The closed loop formed by one of these hydrogen bonds and the intervening stretch of backbone contains 13 atoms (including the hydrogen), as illustrated in Fig. 12. In the usual nomenclature for describing the basic structure of polypeptide helices, the α-helix is known as the 3.613-helix, where 3.6 is the number of residues per turn and 13 is the number of atoms in the hydrogen-bonded loop. The rise per residue along the helix axis is 1.5Å.
FIG. 12. Illustration of the 13-atom hydrogen-bonded loop which determines the subscript in the description of the α-helix as a 3.613-helix (the 3.6 refers to the number of residues per turn). The 13 atoms are those in the shortest covalently connected path which joins the ends of a single hydrogen bond (the hydrogen is one of the 13 atoms): . . . O—C—N—Cα—C—N—Cα—C—N—Cα—C—N—H . . .
The α-helix received strong experimental support when Perutz () found the predicted 1.5 Å X-ray reflection from hemoglobin crystals and from tilted fibers of keratins. The final conclusive demonstration of the α-helix in globular protein structure came from the high-resolution X-ray structure of myoglobin (). It was shown that the myoglobin helices matched Pauling's calculated structure quite closely, and also that they were all right-handed (for L-amino acids, the left-handed α-helix has a close approach between the carbonyl oxygen and the β-carbon). It is easy to determine that, for instance, Fig. 11 is right-handed: if the curled fingers of the right hand are turned in the direction of their tips (as if tightening a screw) and the whole hand is moved in the direction of the outstretched thumb, then a right-handed helical path is traced out. Handedness is an enormously influential parameter in protein structure; most features for which handedness can be defined prefer one sense to the other, and the α-helix is only the first of many examples we will encounter.
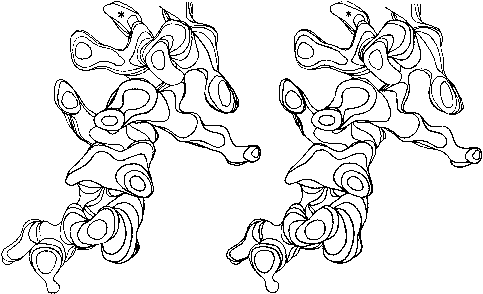
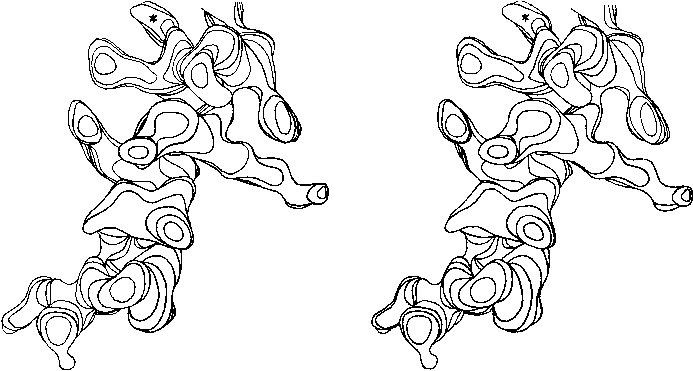
FIG. 13. Stereo drawing of one contour level in the electron density map at 2Å resolution for the residue 54-68 helix in staphylococcal nuclease. Carbonyl groups point up, in the C-terminal direction of the chain; the asterisk denotes a solvent peak bound to a carbonyl oxygen in the last turn. Side chains on the left (including a phenylalanine and a methionine) are in the hydrophobic interior, while those on the right (including an ordered lysine) are exposed to solvent.
Figure 13 shows the electron density map at 2 Å resolution for one of the α-helices in staphylococcal nuclease. Bumps for the carbonyl oxygens are clearly visible; they point toward the C-terminal end of the helix, and are tipped very slightly outward away from the helix axis. At the top, in the last turn of the helix, there is a carbonyl tipped still further outward and hydrogen-bonded to a solvent molecule (marked with an asterisk). Side chain atoms or waters frequently bond to free backbone positions in the first or last turn of a helix, and hydrogen bonds with water are even more favorable for carbonyls than for NH groups (see Section II,H).
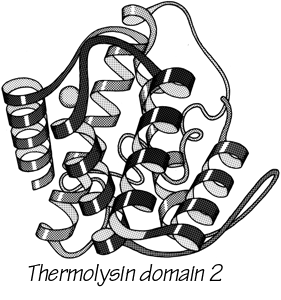
FIG. 14. Schematic drawing of the backbone of an all-helical tertiary structure: domain 2 of thermolysin.
With 3.6 residues per turn, side chains protrude from the α-helix at about every 100° in azimuth. Since the commonest location for a helix is along the outside of the protein, there is a tendency for side chains to change from hydrophobic to hydrophilic with a periodicity of three to four residues (). This trend can sometimes be seen in the sequence, but it is not strong enough for reliable prediction by itself. Different residues have weak but definite preferences either for or against being in α-helix: Ala, Glu, Leu, and Met are good helix formers while Pro, Gly, Tyr, and Ser are very poor (). α-Helices were central to all the early attempts to predict secondary structure from amino acid sequence (e.g., ; ; ; ; ; ; ) and they are still the feature that can be predicted with greatest accuracy (e.g., ; ; ; ; ; ; ). [Helix predictions have now reached better than 70% accuracy, using algorithms such as neural nets or hidden Markov models .] As much as 80% of a structure can be helical, and only seven proteins are known that have no helix whatsoever. Figure 14 shows the second domain of thermolysin, a structure that is predominantly α-helical.
[There are of course now many more than 7 proteins known to have no helicies, but they are still a very small fraction of the total. Further information about amino-acid roles in helix formation is obtained from tabulating position-specific residue preferences . This shows that the ends of helices are very different from the central parts, as described below.]
FIG. 15. Stereo drawing of a bent helix (glyceraldehyde-phosphate dehydrogenase residues 146-161) with an internal proline. The proline ring produces steric hindrance to the straight α-helical conformation as well as having no NH group available for a hydrogen bond. A proline is the commonest way of producing a bend within a single helix, as well as occurring very frequently at the corners between helices.
The backbone conformational angles for right-handed α-helix are approximately φ= -60°, ψ= -60° [, more accurately, -63°, -43°] , which is in a favorable and relatively steep energy minimum for local conformation, even ignoring the hydrogen bonds. α-Helices are certainly the most regular pieces of structure to be found in globular proteins, but even so they show significant imperfections. There can be slight bends in the axis of a helix, of any amount from almost undetectable up to about 20° [30°] (e.g., ), either with or without a break in the pattern of hydrogen bonding. One of the most obvious ways to produce such a bend is with a proline. Proline fits very well in the first turn of an α-helix [especially in position N1] but anywhere further on it not only is missing the hydrogen bond donor but also provides steric hindrance to the normal conformation. It is rare but certainly not unknown in such a position (see Fig. 15). An α-helix is almost invariably made up of a single, connected stretch of backbone (as opposed, for instance, to the backbone changeovers seen for double-helix in transfer RNAs: ). Almost the only known exception to this rule is the interrupted helix from subtilisin that is shown in Fig. 16.
FIG. 16. An unusual interrupted helix from subtilisin (residues 62-86), in which the helical hydrogen bonds continue to a final turn that is formed by a separate piece of main chain. Such interrupted helices (broken on one side of the double helix) are apparently a fundamental feature of nucleic acid structure as illustrated by tRNA, but are exceedingly rare in protein structure.
[It has continued to prove true that strand changeovers are quite common in RNA molecules, with maintenance of base stacking and double helix geometry across the change, but interrupted α-helices with continuous H-bonding across the break remain extremely rare; one further example is in cytochrome C3 (1WAD 74-82), shown in the kinemage II.A_intHlx.kin.]
Click the image to download the ' II.A_intHlx.kin ' kinemage file to view it in interactive 3D with the KiNG software.
The generally regular, repeating conformation in the α-helix places all of the charge dipoles of the peptides pointing in the same direction along the helix axis (positive toward the N-terminal end). It has been shown () that the overall effect is indeed a significant net dipole for the helix, in spite of shielding effects. The helix dipole may contribute to the binding of charged species to the protein: for example, negative nucleotide phosphates, which are typically found near the N-termini of helices.
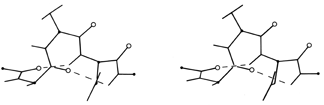
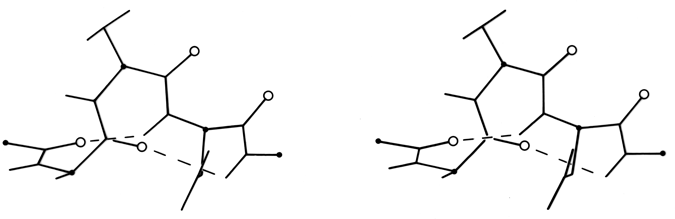
FIG. 17. A short segment of 310 helix from carbonic anhydrase (residues 159-164). Main chain carbonyl oxygens are shown as open circles.
The only other principal helical species besides the α-helix which occurs to any great extent in globular protein structure is the 310-helix (see Fig. 17), with a three-residue repeat and a hydrogen bond to residue n + 3 instead of n + 4. Its backbone conformational angles are approximately φ = -60°, ψ = -30°, [-70°, -20°] , within the same energy minimum as the α-helix. However, for a long periodic structure the 310-helix is considerably less favorable than the α-helix in both local conformational energy and hydrogen bond configuration. In the refinement of rubredoxin at 1.2 Å resolution, Watenpaugh et al. () found that bond angles along the main chain were significantly distorted in all four of the regions that have two successive 310-type hydrogen bonds. Long 310 helices are very rare but short pieces of approximate 310-helix occur fairly frequently. Two consecutive residues in 310 conformation form a good tight turn (see Section II,C), and three consecutive 310 residues forming two interlocked tight turns is also fairly common. But another important location for short bits of 310-helix is at the C-terminal end of an α-helix. It is quite common for the last helical turn to tighten up, with hydrogen bonds back to residue n - 3 or else bifurcated hydrogen bonds to both n - 3 and n - 4 (e.g., ). Némethy et al. (1967) showed that this arrangement is not necessarily quite like 310-helix; they described the αII-helix for this sort of position, which retains the helical parameters of an α-helix but tilts the peptide so that the NH points more inward toward the helix axis and at the same time points more toward the n - 3 than the n - 4 carbonyl. The conformations in real proteins show somewhat of a mixture between the αII tilt and the 310 tightening. Figure 18 shows an example. 310 or αII conformation does not tend to occur nearly as often at the N-termini of α-helices. The reason is that the tighter loop with n + 3-type hydrogen bonds requires the group involved to move closer to the helix axis, either by tilting (αII) or by tightening the helix(310). This motion is easy for the NH group but not for the CO: neighboring carbonyl oxygens would come too close together. [Less often, the end of a helix can loosen rather than tighten or a turn can widen to provide the right geometry for a metal ligand, using the (n+5) H-bonds of what is called a π-helix. An example is myohemerythrin 106-112 (2MHR).]
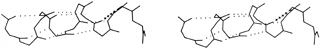
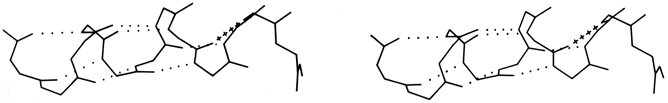
FIG. 18. An example of the αII conformation at the end of the A helix in myoglobin (residues 8-17). The normal α-helical hydrogen bonds are shown dotted, while the tighter αII bond is shown by crosses.
Another frequent feature of the C-termini of helices is a residue (usually glycine) in left-handed α conformation with its NH making a hydrogen bond to the CO of residue n - 5 (see ); this often follows a residue with the 310 or αII bonding described above. [This arrangement has turned out to be very much the commonest way of ending an α-helix. The starting and ending residues that form the transition point half-in and half-out of a helix are now called the helix N-cap and C-cap respectively (Richardson, 1988). The C-cap is most often a glycine in L-α conformation that turns the backbone in the other direction; the peptides NH's on either side of the Gly Cα make H-bonds back to exposed CO's in the last helical turn, but in inverted sequence order (as shown in kinemage II.A_hlxCaps.kin for the Gly C-cap of helix 4 in 1LMB λ repressor). Helix N-cap residues usually have a short sidechain (Asn, Asp, Ser, or Thr) with an oxygen that can H-bond to the exposed backbone NH of residue N2 or N3 (that is, 2 or 3 past the N-cap) in the first helical turn (shown in kinemage for the N-caps of helices 1 and 2 of 1LMB λ repressor). A classic helix N-cap also has a "capbox" reciprocal H-bond from the sidechain of residue N3 (Gln, Glu, Ser, or Thr) to the backbone NH of the N-cap residue, in the peptide just before the start of helical conformation (). Both N-caps and C-caps often also have a "hydrophobic staple" interaction between suitable sidechains at N' and N4 or C4 and C' (). Proline is actually preferred in the N1 positions (Richardson, 1988).
Click the image to download the ' II.A_hlxCaps.kin ' kinemage file to view it in interactive 3D with the KiNG software.
Good N-caps stabilize both entire proteins (; ) and isolated helical peptides (). Glycine C-caps do not stabilize helical peptides (), but that has been shown to be due to their location at the C-terminus of the chain (). Sequences that form good helix caps have become important tools in secondary-structure prediction () and in protein design ().]
A few other helical conformations occur occasionally in globular protein structures. The polyproline helix, of the same sort as one strand out of a collagen structure, has been found in pancreatic trypsin inhibitor () and in cytochrome c551 (). An extended "ε helix" has been described as occurring in chymotrypsin (). In view of the usual variability and irregularity seen in local protein conformation it is unclear that either of these last two helix types is reliably distinguishable from simply an isolated extended strand; however, the presence of prolines can justify the designation of polyproline helix.
[sidebar: Analyses of Helix-Helix Packing]
***The ways in which α-helices pack against one another were initially described by Crick () as "knobs into holes" side chain packing which could work at either a shallow left-handed crossing angle or a steeper right-handed one. Helix-helix interactions have recently been analyzed in more detail by several different groups, using quite varied approaches and points of view. Chothia et al. () considered the helix contact angles at which ridges formed by rows either of n,n + 3 or of n,n + 4 side chains can pack against each other. They predict three classes (I, II, and III) of contact at angles of -82°, -60° and +19°, respectively (the angle is handed but does not consider direction of the helices). For 25 cases they find a distribution consistent with these classes, although there is better discrimination between classes II and III than between I and II. Richmond and Richards () determine contact residues by calculating solvent accessible area lost on bringing helix pairs together, and model the interactions using helices of close-packed spheres. They find contact classes that match the packing of Chothia's classes II and III, but for approximately perpendicular helices (class I) they find a favorable contact only if the two central residues are glycine or alanine and pack directly on top of each other. In globins the helix axes are about 2Å closer together for steeply angled contacts than for nearly parallel ones, which have a long contact surface between relatively large residues. Figure 19 shows stereo drawings of class II and class III helix contacts. Efimov (, ) also considers side chain packing as the determinant for helix contacts, but from a rather different theoretical perspective. He first considers what side chain conformations will allow close packing of neighboring hydrophobic side chains on a single helix, then considers how to close-pack side chains of hydrophobic patches on the buried side of two parallel or antiparallel helices, then finally considers the angles for packing together two layers of helices by matching two of the relatively flat hydrophobic surfaces produced in the second step.
Each of these approaches has its advantages; the contact nets drawn by Chothia et al. are the only version that explicitly shows the actual (rather than idealized) residue contacts, but they have made correlations only with the one variable of contact angle. Efimov has obtained a very interesting regularity that successfully predicts side chain conformation at the right and left edges of hydrophobic strips, but has not considered either the interactions directly in between helix pairs in his first step or the possibility that close (as opposed to distant hydrophobic) contacts could occur at steep angles. Richmond and Richards have the advantage of identifying residue contacts in a way that is not influenced by theoretical preconceptions, and they have considered side chain identity (although not conformation) in detail. Because of the great local variability of side chain size and packing and because relatively few examples have yet been analyzed, it is obviously possible to describe a given contact as fitting quite different idealized models. The current large data set of proteins shows a strong tendency for class III (shallow) interactions to be antiparallel and for parallel helix interactions to be class II. It seems likely that the antiparallel up and down helix bundle structures (see Section III,B) would be composed of paradigm class III interactions, and the doubly wound α/β structures (see Section III,C) would contain paradigm class II interactions, but none of the 15 proteins analyzed by the above three methods happen to fall into either of those categories. If multiple examples of paradigm classes II and III contacts can be analyzed and compared, it may then be possible to define a meaningfully distinct class of perpendicular contacts.***
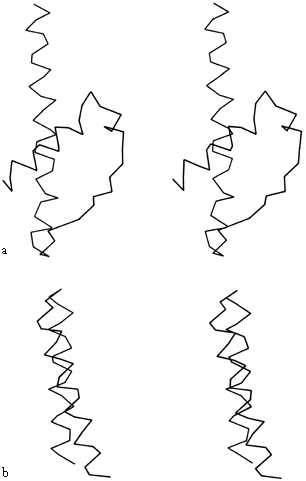
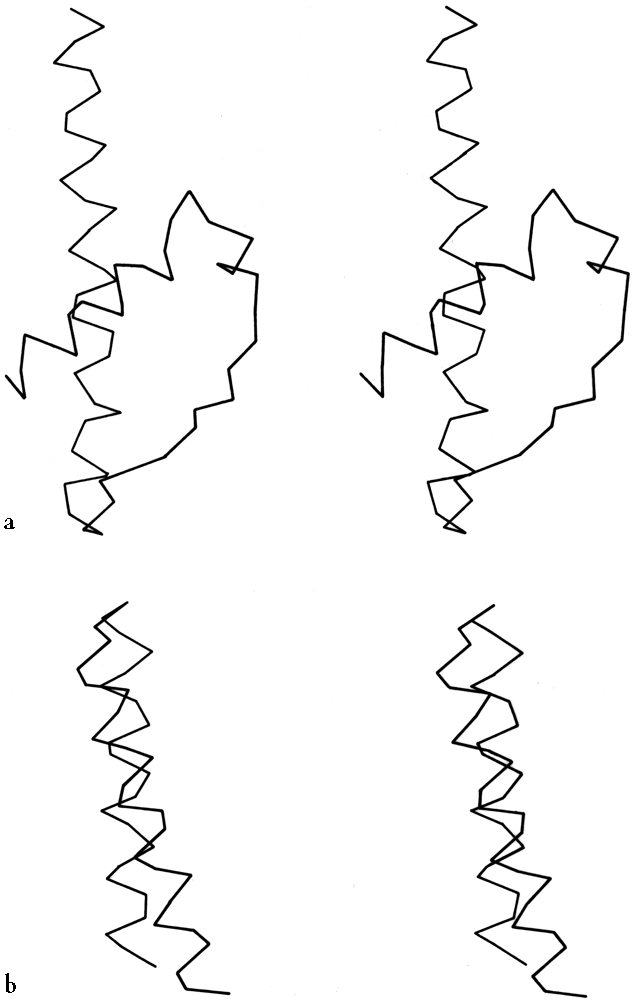
FIG. 19. Examples of the two commonest types of helix-helix contact: (a) Class II (from hexokinase) with an inter-helix angle of about -60°; (b) Class III (from myohemerythrin) with an inter-helix angle of about +20°.
[Analysis of the general geometry of helix packing is still a fairly open issue, but several aspects of the problem have progressed. An interesting treatment of helix packing in terms of alternate edges of polyhedra () fits well for many but not all structures. The common and biologically important case of coiled-coils (belonging to the low-angle, class III case) has been very thoroughly and successfully described (), and a rare type of low-angle contact at much closer distances has been described (). Perpendicular T-junction contacts have proven important in Ca++-binding "E-F hands" () and DNA-binding helix-turn-helix motifs ().]