
The other major structural element found in globular proteins is the β sheet. Historically, it was first observed as the β, or extended, form of keratin fibers. An approximate understanding of the molecular structure involved was achieved much earlier for the β than for the α structure, because repeat distances along the fiber showed that the backbone must be almost fully extended, which did not leave very much choice of conformation even when the details of backbone geometry were not well known. Astbury described the β structure in 1933 as straight, extended chains with alternating side chain direction and hydrogen bonds between adjacent antiparallel chains. Pauling and Corey () described the correct hydrogen-bonding patterns for both antiparallel and parallel β sheet, and also realized that the sheets were "pleated," with α-carbons successively a little above and below the plane of the sheet. Some features of β structure, such as its characteristic twist, were not recognized until after several β sheets had been seen in three-dimensional protein structures.
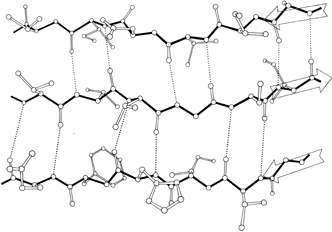
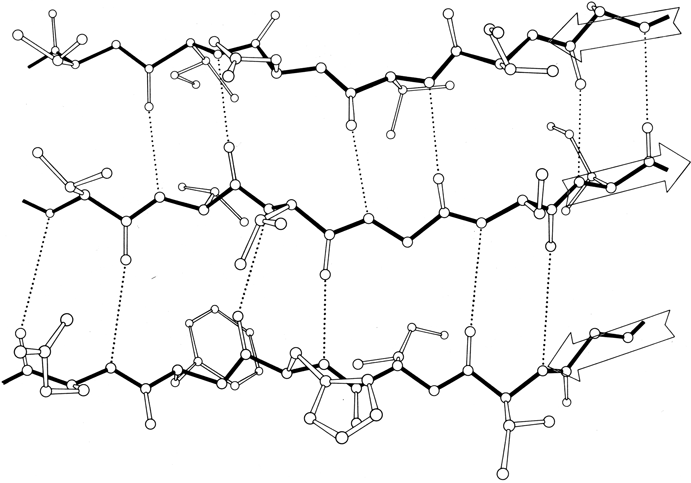
FIG. 20. An example of antiparallel β sheet, from Cu,Zn superoxide dismutase (residues 93-98, 28-33, and 16-21). Arrows show the direction of the chain on each strand. Main chain bonds are shown solid and hydrogen bonds are dotted. In the pattern characteristic of antiparallel β sheet, pairs of closely spaced hydrogen bonds alternate with widely spaced ones. The direction of view is from the solvent, so that side chains pointing up are predominantly hydrophilic and those pointing down are predominantly hydrophobic.
β sheet is made up of almost fully extended strands, with φ,ψ angles which fall within the wide, shallow energy minimum in the upper left quadrant of the Ramachandran plot (see Figs. 7 and 9). β strands can interact in either parallel or antiparallel orientation, and each of the two forms has a distinctive pattern of hydrogen bonding. Figures 20 and 21 illustrate examples of antiparallel and parallel β sheets from real protein structures. The antiparallel sheet has hydrogen bonds perpendicular to the strands, and narrowly spaced bond pairs alternate with widely spaced pairs. Looking from the N- to C-terminal direction along the strand, when the side chain points up the narrow pair of H bonds will point to the right. Parallel sheet has evenly spaced hydrogen bonds which angle across between the strands.
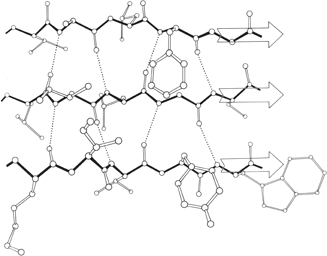
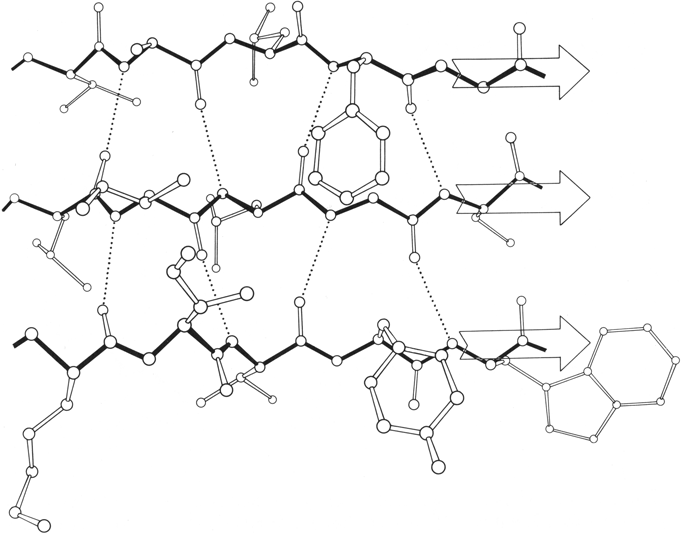
FIG. 21. An example of parallel β sheet, from flavodoxin (residues 82-86, 49-53, and 2-6). In the pattern characteristic of parallel β sheet, the hydrogen bonds are evenly spaced but slanted in alternate directions. Since both sides of the sheet are covered by other main chain (as is almost always true for parallel sheet), side groups pointing in both directions are predominantly hydrophobic except at the ends of the strands.
Within a β sheet, as within an α-helix, all possible backbone hydrogen bonds are formed. In both parallel and antiparallel β sheet, the side groups along each strand alternate above and below the sheet, while side groups opposite one another on neighboring strands extend to the same side of the sheet and are quite close together. These close side chain pairs on neighboring strands show preferences for having hydrophobic groups together, unlike charges together, and branched β-carbons next to unbranched β-carbons (in antiparallel sheet), but none of these preferences are stronger than 2 to 1. Lifson and Sander () have shown that specific residue pairs on neighboring strands recognize each other, over and above simple grouping by polarity, but again they comment on the fact that the correlations are not as strong as one would have expected. As an example of the kind of factors involved, let us examine the interactions of a pair of side chains with branched β-carbons on neighboring strands of β sheet. Valine and isoleucine have a rather strong conformational preference (better than two-thirds of the cases) for the χ1 orientation staggered relative to the main chain (). Since the relation between adjacent parallel strands is a translation, neighboring Val or Ile residues in the preferred conformation "cup" against each other back-to-front in a very favorable packing. Since the relationship between adjacent antiparallel strands is twofold, in that case a pair of side chains with the preferred χ1 angle will either pack back-to-back leaving unfilled space or else front-to-front, which produces a collision unless the main chain conformation is adjusted. The effects of these restrictions can indeed be seen in the patterns of residue-pair occurrence, but only weakly. Looking at the actual pairs of, for instance, Val-Val or Val-Ile in antiparallel sheet, one finds either that one of the side chains has adopted an unfavorable χ1 angle so that the two can pack well (as in the upper left corner of Fig. 20) or else the main chain has twisted to put the β-carbons at an optimum distance (e.g., when a Leu-Val pair in chymotrypsin becomes a Val-Val pair in elastase, the β-carbons move 0.65 Å further apart). This in turn, of course, shows one reason why the χ1 preference is not stronger or the φ,ψ angles more regular. In general, the impression one takes away from this kind of examination is that the protein is balancing so many factors at the same time that there are always ways to compensate for any individual problem. Thus studies of individual parameters uncover only weak regularities in spite of the strength of the overall packing constraints. Looking at long strings of adjacent side chains across the centers of large sheets, such as shown in the stereo figures of Lifson and Sander (), one sees a stronger expression of the packing difference between antiparallel and parallel sheets: Ile-Leu-Val-Leu and Val-Ala-Thr-Gly-Ile in elastase and Ala-Ile-Ala-Val, Ala-Ile-Leu-Ile-Ala, and Ser-Thr-His-Val-Ser in concanavalin A, versus Val-Val-Ile-Val-Val-Val and Ile-Val-Ile in glyceraldehyde-phosphate dehydrogenase domain 1 and Val-Val-Ile, Val-Val-Val, and Ile-Ile-Val in triosephosphate isomerase. [Wouters and Curmi (1995) give an updated statistical study of pair frequencies in β sheet, while the energetics of specific replacements have been studied experimentally, especially in the nicely behaved B domain of protein G (e.g., Smith et al. 1994).]
β strands can combine into either a pure parallel sheet, a pure antiparallel sheet, or a mixed sheet with some strand pairs parallel and some antiparallel. If the assortment were random there would be very few pure sheets, but in fact there is a strong bias against mixed sheets (), perhaps because the two types of hydrogen bonding need slightly different peptide orientations. Only about 20% of the strands inside β sheets have parallel bonding on one side and antiparallel on the other.
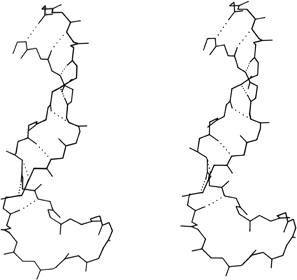
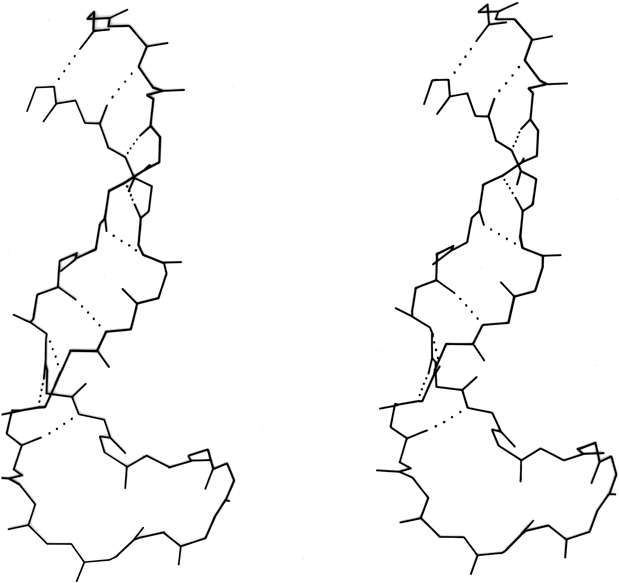
FIG. 22. An example of a long two-stranded ribbon of antiparallel β structure, from lactate dehydrogenase (residues 263-294). Side chains are not shown; hydrogen bonds are dotted. As is typical of isolated two-stranded ribbons, the chains show a very strong twist (180° in about five residues).
Parallel β sheet is in general a good deal more regular than antiparallel. If φ,ψ angles are plotted for both types of sheet, as for instance in Nagano (), the parallel residues cluster rather tightly while the antiparallel ones spread over the entire quadrant. Parallel β structure almost never occurs in sheets of less than five total strands, whereas antiparallel β structure often occurs as a twisted ribbon of just two strands. Figure 22 shows such a two-stranded antiparallel β ribbon. Parallel β sheets and the parallel portions of mixed sheets are always thoroughly buried, with other main chain (often α-helices) protecting them on both sides. Antiparallel sheets, on the other hand, typically have one side exposed to solvent and the other side buried, so that they often show an alternation of side chain hydrophobicity in the amino acid sequence. [The contrast is still very real, but there are now examples of solvent-exposed parallel β sheet: on some parts of parallel β-helix structures such as LpxA (1LXA), or on the inner surface of α/β horseshoes such as ribonuclease inhibitor (1DFJ). This seems to happen, however, only for highly repetitive and very regular parallel β-sheets, and probably benefit from stabilization by their cooperativity.] β sheets in general show a tendency toward greater hydrophobicity for the central than for the edge strands of the sheet (). These three requirements of parallel β sheets (regularity, size, and protection) all suggest that parallel β structure is less stable than antiparallel (), since it apparently needs the cooperativity of an extensive hydrogen-bond network () and also seems to need those hydrogen bonds shielded from water. (It is actually possible to shield the backbone with large hydrophobic side chains, but those are not the residues that would occur on an exposed surface.) Mixed β sheets tend to have the general appearance characteristic of their predominant H-bonding type. Sheets that are approximately half and half, such as carboxypeptidase or carbonic anhydrase, tend to look like parallel sheets because they require substantial protection on both sides. Figure 23 is a schematic drawing of a typical parallel-type β sheet structure in a protein.


FIG. 23. Schematic drawing of the backbone of flavodoxin, a protein in which a parallel β sheet is the dominant structural feature. The sheet (represented by arrows) is shown from one edge, so that the characteristic twist can be seen clearly.
One of the most conspicuous features of β sheet as it occurs in the known protein structures is its twist (). This twist always has the same handedness, although it has unfortunately been described by two conflicting conventions in the literature. If defined in terms of the angle at which neighboring β strands cross each other, then the twist is left-handed (e.g., ; ); if defined in terms of the twist of the hydrogen bonding direction or of the peptide planes as viewed along a strand, then the twist is right-handed (e.g., ; ). We will use the right-handed definition in this article, because it is meaningful even for an isolated strand. [The right-handed definition won out and is now standard.] Figure 23 shows the side view of a β sheet in which the twist is obvious.
There is of course no a priori reason to expect the flat n = 2 conformation to be especially favored for handed amino acids; however, the exact mechanism by which L-amino acids favor right-handed strand twist is not entirely obvious and has been explained in several different ways. Detailed calculations of local conformational energy (e.g., ) always place the minimum well off to the right of the n = 2 line of a flat strand (see ) although the minimum is a very broad, shallow one. Chothia () points out that probabilistic effects will produce a right-handed average twist, since many more of the accessible conformations within the general β area on the φ,ψ plot lie to the right of the n = 2 line. Raghavendra and Sasisekharan () have found that inclusion of H bond and nonbonded interactions between a pair of antiparallel β strands produces a considerably deeper calculated energy minimum in the right-handed region. There is some evidence from small-molecule peptide crystal structures () of a systematic tetrahedral distortion at the peptide nitrogen, and Weatherford and Salemme () have shown that the combination of that distortion with optimal β sheet hydrogen bond geometry would favor a right-handed strand twist. In the known structures, β strand twist varies from close to 0° per residue to about 30° per residue, with the highest values for two-stranded ribbons (see Fig. 22) and generally lower values the more strands are present and the longer they are. This indicates some degree of conflict between the requirements for optimal hydrogen bonding and for lowest local conformational energy. [Indeed, if a large sheet were strongly twisted then the H-bonds would have to be longer at the edges; a greater degree of "pleat" at the sheet center helps somewhat, but flatness helps even more. These relationships were explored in Salemme ().]

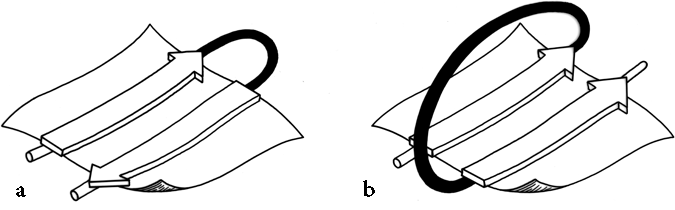
FIG. 24. The two major sorts of connection between β strands: (a) a "hairpin," or same-end, connection (this example is type +1, to a nearest-neighbor strand); (b) a "crossover," or opposite-end, connection (this one is type + lx).
Once it has been decided what β strands belong in a given sheet (a process involving occasional subjective decisions for marginal cases), then it is possible to give a simple and unambiguous description of the topological connectivity of those strands in the sheet (). Each connection between two β strands must fall into one of two basic categories: hairpin connections in which the backbone chain reenters the same end of the β sheet it left, and "crossover" connections in which the chain loops around to reenter the sheet on the opposite end (see Fig. 24). Each connection is named according to how many strands it moves over in the sheet and in which direction, with an "x" added for crossover connections. Thus, a "+1" is a hairpin and a "+1x" a crossover connection between nearest-neighbor strands; a "+2" is a hairpin and a "+2x" is a crossover connection that skips past one intervening strand in the sheet, and so on. The conformation of the connecting loop is irrelevant to this topological designation. Nearest-neighbor connections of +1 and +1x are by far the most common, occurring about three times as frequently as all other connection types put together ( ).
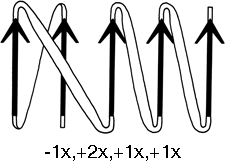
FIG. 25. A topological schematic diagram of the connectivity in the parallel β sheet of flavodoxin. Arrows represent the β strands; thin-line connections lie below the plane of the sheet and fat connections above it. No attempt is made to indicate the length or conformation of the connecting chains (most of them are helical) or the twist of the β sheet. The topology can also be specified by a sequential list of the connection types: in this case, -lx, +2x, +1x, +1x.
The topology of an n-stranded β sheet can be specified by a list of its n-1 connections, starting from the N-terminus. For example, flavodoxin (Fig. 23) can be described as either +1x, -2x, -1x, -1x, or -1x, +2x, +1x, +1x (absolute value of the signs is not meaningful, since the sheet could be turned upside down). We will use connection types to describe and classify β sheets, and will also use a simplified kind of topology diagram (see Fig. 25) which views the sheet from above. There is another type of topology diagram also common in the literature which views the sheet end-on (see ); the topology is less explicit but more features of the three dimensional structure are retained. That is a significant advantage in the cases in which it works best, but since adherence to the convention forces substantial distortions in some proteins, we will use separate diagrams for the three-dimensional structure and for the topology in the overall survey (see Sections III,A-E).
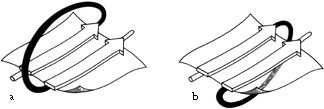
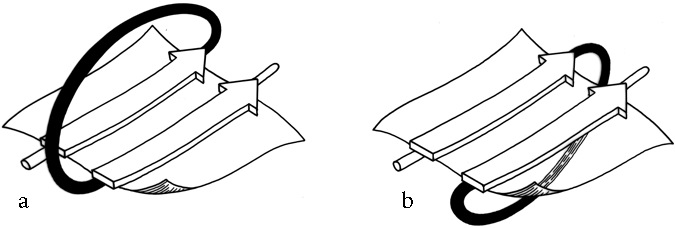
FIG. 26. (a) A right-handed +1x crossover connection; (b) a left-handed +1x crossover connection.
Crossover connections have a handedness (see Fig. 26), since they form a loose helical turn from one strand, up (or down) and around, and back into the next strand. Essentially every one of the crossover connections in the known protein structures regardless of the length or conformation of the connecting loop, is right-handed ( ). There is one really well authenticated left-handed crossover in subtilisin and one in glucose-phosphate isomerase in a region where the chain connectivity is not completely certain (), while there are many more than a hundred right-handed crossovers. [The enormous preference for right- over left-handed crossover connections has held true.]
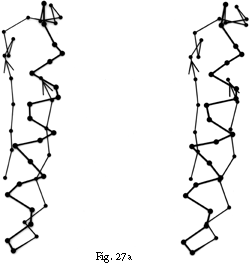
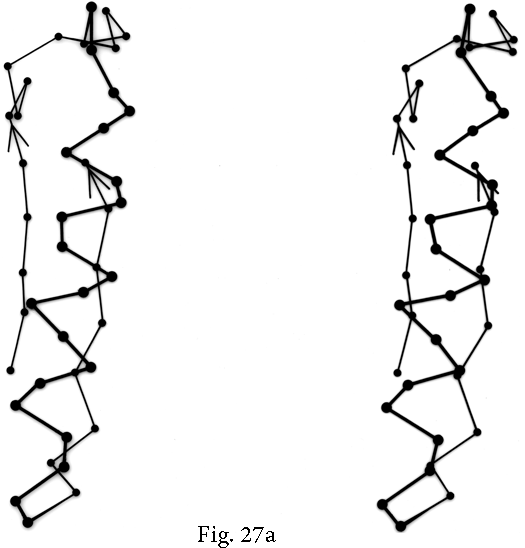
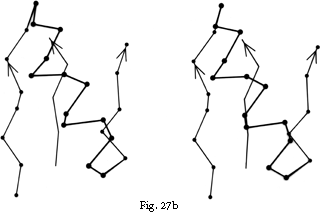
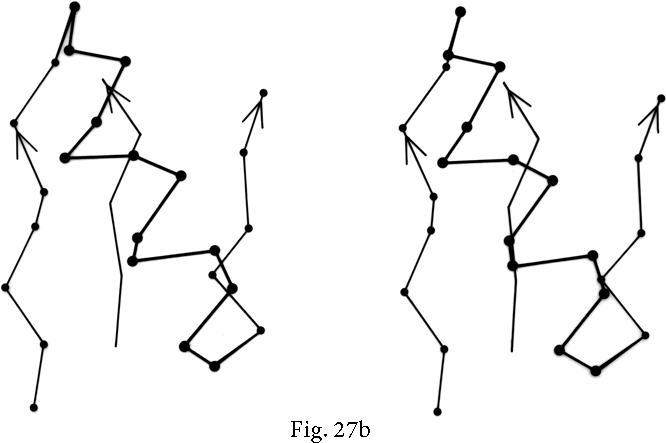
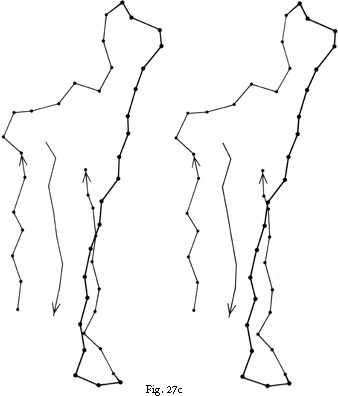
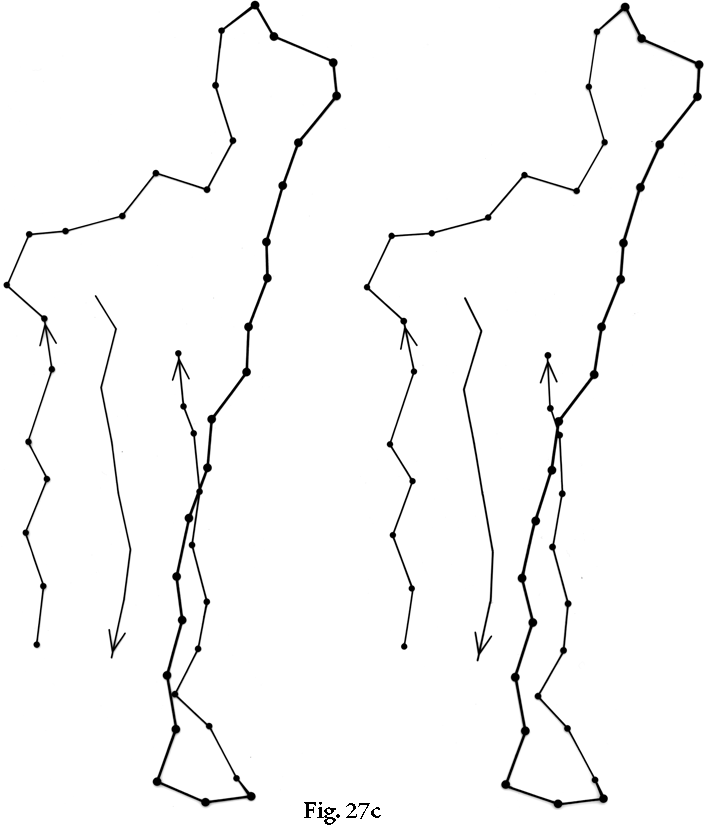
FIG. 27. Examples of particular crossover connections: (a) a right-handed +1x, residues 200-242 from carboxypeptidase A; (b) a right-handed +2x, residues 109-133 from papain; (c) a right-handed +2x, residues 169-214 from concanavalin A.
Over half of the crossover connections have at least one helix in the connecting strand, and in many of those cases the helix packs against one or both of the β strands it connects (see Fig. 27a). Sternberg and Thornton () have explained the handedness by the fact that β sheet twist makes the right-handed connection shorter and more compact (as can be seen in Fig. 26). Nagano () has explained the handedness by the preferred packing angles of a helix against a β strand, which again would allow more compact and shorter corners (between the α and β elements) in the right-handed form. Both of these explanations are sure to be important contributing causes of crossover handedness, but they are limited to the relatively short, straightforward examples with tight corners. The large number of crossover connections which are too long, start off in the wrong direction, or do not pack against the β sheet (see Fig. 27a and c for examples) show almost as strong a handedness constraint as the more classic cases. In an attempt to account for these long examples, Richardson () proposed a hypothetical folding scheme for crossover connections by which the twist of a long extended strand or of a helix flanked by extended chains is transferred to the crossover loop as the backbone curls up (see Fig. 28). However it is achieved, the right-handedness of crossover connections is the dominant factor controlling the appearance of both singly-wound and doubly-wound parallel α/β structures (see Section III,C). Crossover connections are also fairly common in antiparallel β sheet.
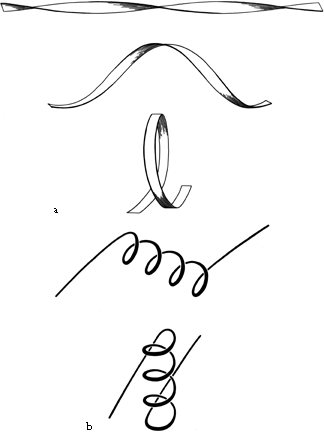
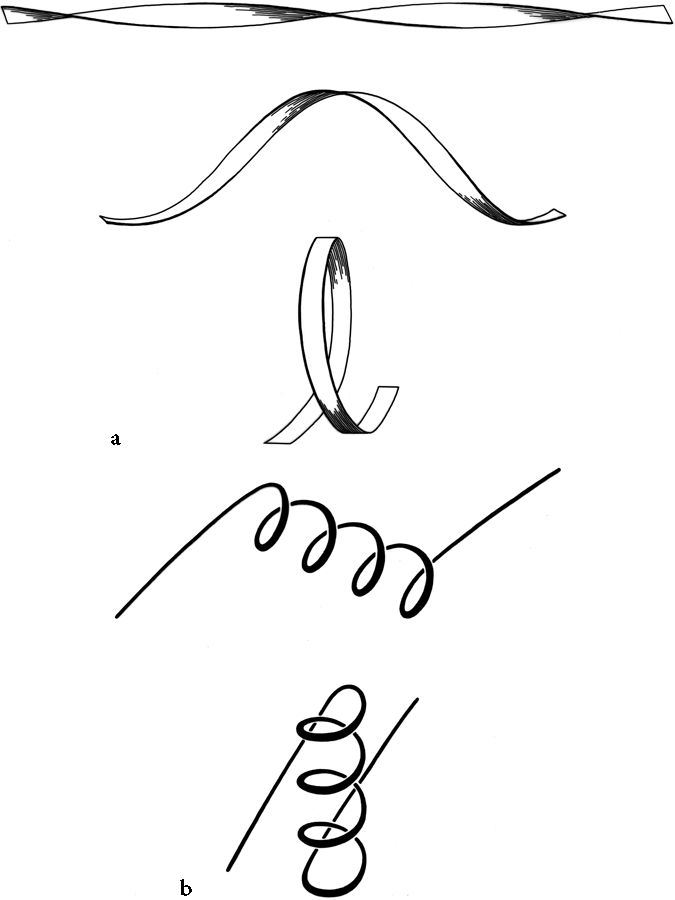
FIG. 28. Illustration of possible folding schemes which would produce the handedness of crossover connections as a consequence of (a) the handedness of twist of an initial β ribbon, or (b) the handedness of an initial α-helix.
Parallel β structure usually forms large, moderately twisted sheets such as in Fig. 23, although occasionally it rolls up into a cylinder with helices around the outside (e.g., triosephosphate isomerase). Large antiparallel sheets, on the other hand, usually roll up either partially (as in the first domain of thermolysin or in ribonuclease) or completely around to join edges into a cylinder or "barrel." Occurrence, topology, and classification of β barrels will be discussed in Section III,D, but here we will consider the interaction between the β sheets on opposite sides of the barrel, especially in terms of the angle at which opposite strands cross.
β barrels may be made up of as few as 5 or as many as 13 strands. [Even larger, and often quite round, β-barrels occur in membrane-spanning proteins such as the porins, but their insides are not filled by the β-sheet side chains: they are either open or contain loops.] Their interiors are packed with hydrophobic side chains, which are found to have the same average side chain volume as for a normal amino acid composition. There are no large barrels filled with tryptophans or small ones filled with alanines, presumably because mutation to change the size of even as many as two or three residues at once would still produce a bad fit. The cross sections of all the barrels look remarkably alike, regardless of strand number, with a slight flattening in one direction. Figure 29 shows examples of cross sections from real β barrels with different numbers of strands. The nearly constant appearance is obtained by varying the degree of strand twist around the barrel.
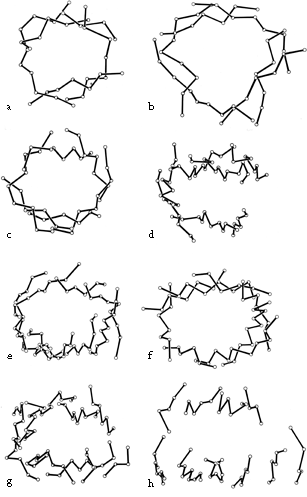
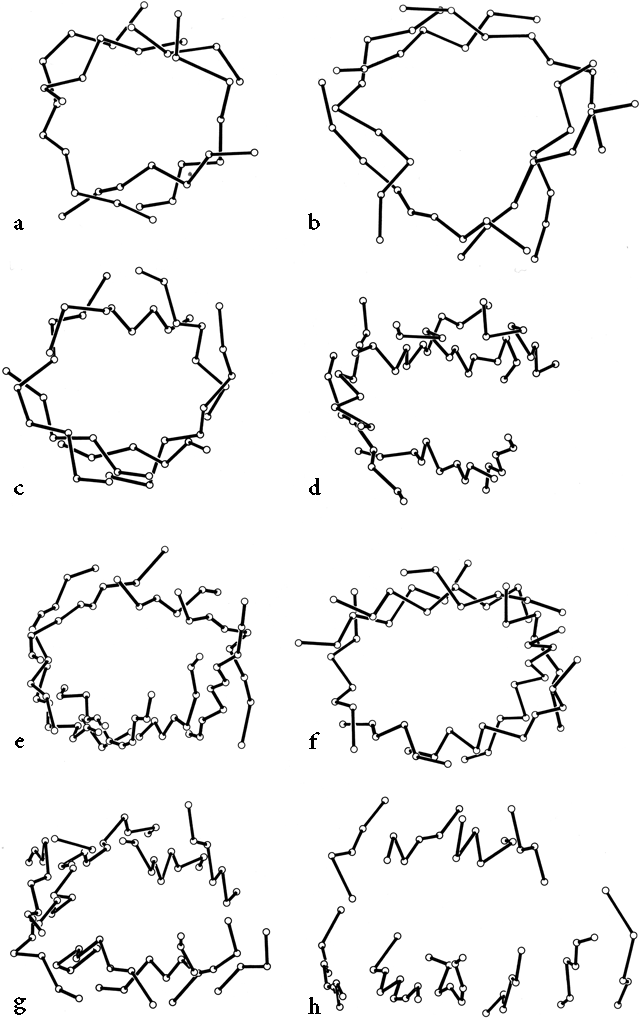
FIG. 29. An assortment of β barrels, viewed down the barrel axis: (a) staphylococcal nuclease, 5-stranded; (b) soybean trypsin inhibitor, 6-stranded; (c) chymotrypsin, 6-stranded; (d) immunoglobulin (McPC603 CH1) constant domain, 7-stranded; (e) Cu,Zn superoxide dismutase, 8-stranded; (f) triosephosphate isomerase, 8-stranded; (g) immunoglobulin (McPC603 VH) variable domain, 9-stranded; (h) tomato bushy stunt virus protein domain 3, 10-stranded. Twist decreases significantly as strand number increases, but cross section stays nearly constant.
Like the bias-woven finger-bandages which tighten around a finger when stretched, a barrel with a given number of strands has a smaller diameter the less twist it has. Twist can be measured by the angle at which strands on opposite sides of the barrel cross one another; that angle averages 95° for 5- and 6-stranded antiparallel barrels, 40° for 7- and 8-stranded ones, and 30° for 9- through 13-stranded ones. Barrel diameter can also be maintained with fewer strands by separating one or more strand pairs further apart than normal hydrogen-bonding distance; this is a very pronounced effect in plastocyanin, for instance, which has a very low twist angle for an 8-stranded barrel. Eight-stranded parallel barrels are more twisted (averaging 75°) than 8-stranded antiparallel ones because all of their strands are hydrogen-bonded and more regular. Beyond eight or nine strands the twist cannot decrease any further and the barrel cross section simply flattens more, keeping the same short axis (11-12Å).