
One important reason for classifying proteins is simply to make the structures more memorable. The system proposed above can help to do that, especially for those cases which fall into one of the more narrowly defined subgroups. However, we also want to know to what extent this classification is a true taxonomy: that is, whether or not it expresses underlying genetic relationships. In addition, among so many structure examples, almost any major rule governing either protein evolution or protein folding would have predictable statistical consequences on the pattern of structural resemblances to be expected. Therefore, it is worthwhile examining the distribution of features that is actually found, because it may suggest various conclusions about how proteins evolve and fold.
|
Internal Similarity or Dissimilarity of Domains within Multidomain Proteinsa
|
|
Similar domain pairs |
Different domain pairs |
| Phosphorylase d1, d2 | Papain d1, d2 |
| Phosphoglycerate kinase d1, d2 | Tyrosyl-tRNA synthetase d1, d2 |
| Aspartate carbomoyltransferase catalytic d1, d2 | Thermolysin d1, d2 |
| Arabinose-binding protein d1, d2 | T4 phage lysozyme d1, d2 |
| Phosphofructokinase d1, d2 | Glucosephosphate isomerase d1, d2 |
| Rhodanese d1, d2 | Pyruvate kinase d1, d2 |
| Hexokinase d1, d2 | Pyruvate kinase d2, d3 |
| Glutathione reductase d1, d2 | Lactate dehydrogenase d1, d2 |
| Tomato bushy stunt virus d2, d3 | Alcohol dehydrogenase d1, d2 |
| Chymotrypsin d1, d2 | Glyceraldehyde-phosphate dehydrogenase d1, d2 |
| γ-Crystallin d1, d2 | Glutathione reductase d2, d3 |
| Immunoglobulin C, V | Influenza virus hemagglutinin HA1, HA2 |
| Immunoglobulin C1, C2 | p-Hydroxybenzoate hydroxylase (4-hydroxybenzoate 4-monooxygenase) d1, d2 |
| Immunoglobulin C2, C3 | p-Hydroxybenzoate hydroxylase d2, d3 |
| Acid protease d1, d2 | Catalase d1, d2 |
| Wheat germ agglutinin d1, d2 | Catalase d2, d3 |
| Wheat germ agglutinin d1 and d2, d3 and d4 | |
a For proteins with more than two domains, each potential duplication is listed separately: e.g., a minimum of two duplications would be necessary to produce either a three-domain or a four-domain structure. Members of the pairs in the left-hand column both fall within the same structural subcategory and have fairly similar topologies; such pairs are perhaps the result of internal gene duplications. Members of pairs in the right-hand column almost all fall into different major categories of tertiary structure (e.g., one all-helical and one antiparallel β); presumably they could not have been produced by internal gene duplication. | |
One significant feature evident in the known structures is the frequency with which domain pairs within a given protein are found to match each other closely in structure. It is known from amino acid sequences (e.g., ) that internal gene duplication can occur in proteins. For recent or highly conserved duplications with closely related sequences the duplication event can be conclusively demonstrated. However, study of sequences cannot tell us how widespread and frequent gene duplication has been in the evolution of proteins because it cannot detect old duplications whose sequences have had time to diverge beyond recognizable homology. There are 26 multidomain proteins in our sample, which would have required the introduction of new domains 35 different times; they are listed in Table II. In slightly over half (17) of those cases, the structure of the new and old domains is basically the same (Fig. 106 shows the two domains of rhodanese as an example); in two cases (cytochrome c peroxidase and aspartate transcarbamylase regulatory chain) the level of similarity is ambiguous; while in the other 16 cases the structures are totally different and presumably could not be the result of internal gene duplication (e.g., Fig. 107). Many of the 17 similar cases involve rather unusual structures, such as the complex mixed sheets of the acid proteases, the five-layer domains of phosphorylase or the mixed doubly wound sheets of hexokinase (Fig. 108).
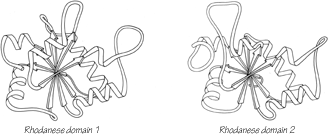
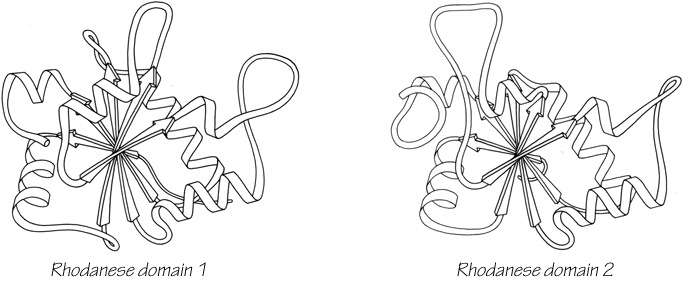
FIG. 106. Rhodanese domains 1 and 2 as an example of a protein with two domains which resemble each other extremely closely.
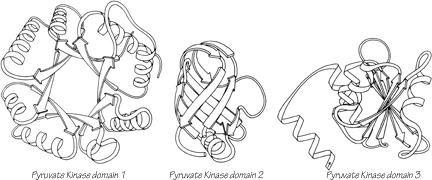
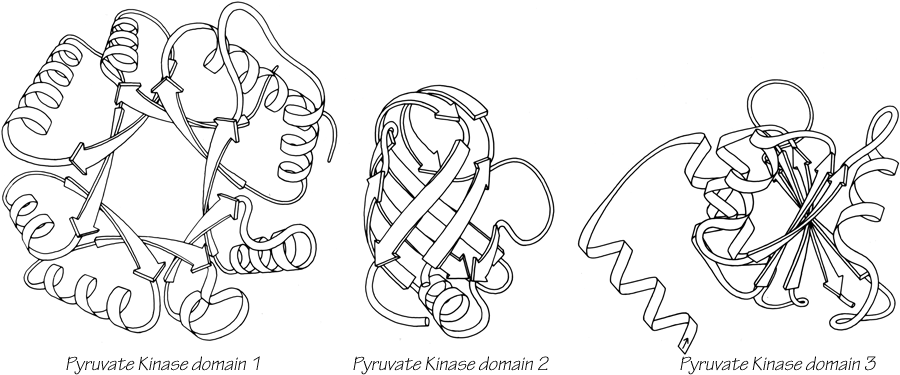
FIG. 107. Pyruvate kinase domains 1, 2, and 3 as an example of a protein whose domains show no structural resemblance whatsoever.
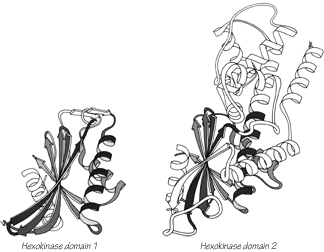
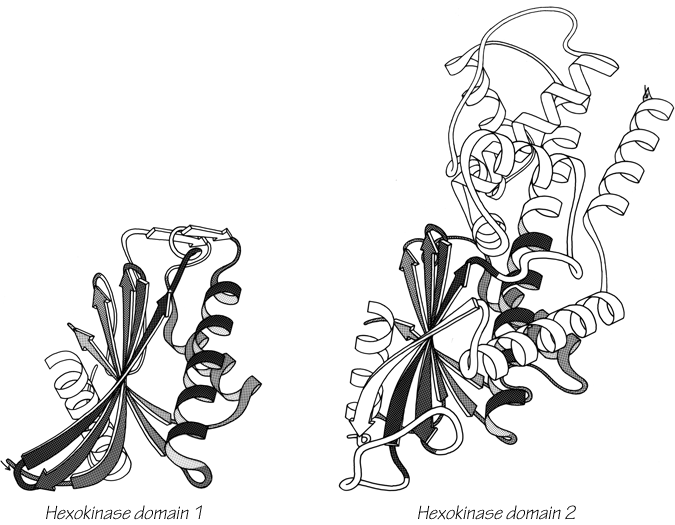
FIG. 108. Hexokinase domains 1 and 2: the proteins whose domains are least alike of all the cases that may represent gene duplications. The equivalent portions of the two domains are shown shaded.
In only 4 of the 17 similar domain pairs is it possible to find a domain in some other protein that matches the structure of one of the pair as well as they match each other (two of those four cases are classic-topology doubly wound sheets). Only for the immunoglobulins (and probably for wheat germ agglutinin, if its sequence were known) is there any significant sequence homology detectable between the similar domain pairs. Purely by chance one would expect vaguely similar structures (within the same major subgroup) perhaps one time out of ten, and detailed resemblance of relatively unusual structures only about one time in 50 or 100. It is unlikely therefore, that more than one or two of the 17 similar domain pairs happened by chance. Only one or two of the pairs could be the products of convergent evolution, because in the other cases the two domains of the pair have quite different functions. Therefore it seems fairly certain that almost all of the similar domain pairs are indeed the result of internal gene duplications. We are left then with the rather interesting conclusion that about half the time multiple domains are produced by gene duplication.
It is also possible that the large and relatively complex domain structures we find today were initially produced by gene duplication from smaller substructures; many of these cases have been analyzed by McLachlan (e.g., ; ). There is very strong evidence from sequence as well as structure that this occurred in ferredoxin () and probably in the carp calcium-binding protein (). These substructures would not have been very stable by themselves, but they could perhaps have survived under less rigorously competitive conditions by associating as identical subunits. (There must have been a stage early in the evolution of life when there were few proteases to degrade a temporarily unfolded protein; also, marginally stable proteins become a selective disadvantage only when other organisms develop more stable ones.) It is very difficult to tell from the present structures whether or not this process commonly occurred. Most such possible substructures are so simple that they are very likely to occur often no matter how the domains originated, which means that the internal symmetry of the doubly wound, singly wound, up-and-down α, and up-and-down β structures does not prove that they originated by duplication Also, such duplications would have to have been extremely ancient genetic events and there could have been much alteration since then, so the fact that most other structures show no internal symmetries does not prove that they did not originate by such a process. For example, the uteroglobin subunit (see Fig. 88) is not internally symmetric because its fourth helix exchanges position with the one on the other subunit of the dimer; it is very unclear, however, that uteroglobin is any less likely to be the product of an internal duplication than, for instance, myohemerythrin.
The next evolutionary question is how many of the different proteins are related to one another. To what extent are the various proteins in one of the structure subgroups all related? To what extent are proteins within a functional category related? In the end, this question comes down to asking how difficult it is to originate completely new proteins: are there very many, or relatively few, independent evolutionary lines among the proteins? At one extreme we would expect to see a fairly small number of distinguishable general structure types, and all members of one functional category of proteins would usually be found within the same structural class. If there were improbable structures represented, as there might well be, there would be only a few different such structure types but each would include several similar protein examples. At the other extreme, we would expect a fairly random distribution of functional types within the various structural categories, with many different improbable structures represented by only a single example of each. Neither of these extreme situations quite applies to the observed distribution of protein structures, but in general it conforms much more closely to the multiple-origin, random model. The simplest, most probable structure types are extremely common, while the more peculiar, complicated patterns each show up only once or twice.
In a very broad overview of the structural categories one can state several statistical correlations with type of function. Hemes are almost always bound by helices, but never in parallel α/β structures. Relatively complex enzymatic functions, especially those involving allosteric control, are occasionally antiparallel β but most often parallel α/β. Binding and receptor proteins are most often antiparallel β, while the proteins that bind in those receptor sites (i.e., hormones, toxins, and enzyme inhibitors) are most apt to be small disulfide-rich structures. However, there are exceptions to all of the above generalizations (such as cytochrome c3 as a nonhelical heme protein or citrate synthase as a helical enzyme), and when one focuses on the really significant level of detail within the active site then the correlation with overall tertiary structure disappears altogether. For almost all of the dozen identifiable groups of functionally similar proteins that are represented by at least two known protein structures, there are at least two and sometimes four or five totally different tertiary-structure types which share that function, as shown in Table III. Probably the most dramatic case is the proteolytic enzymes: although the trypsin-like serine proteases form a structurally related group, the proteases as a whole are represented by six widely different structures, including two textbook examples of convergent evolution: subtilisin versus the trypsin family and thermolysin versus carboxypeptidase. Even in the cases in which only two protein structures are known from a general functional category (such as lectins, nucleases, peroxidases, oxygen carriers, etc.) those two structures are quite different. It seems possible, then, that active sites are easier to alter or to redevelop independently than one would have thought, compared with the total time scale involved in the evolution of proteins.
|
Correlation between Functional Descriptions of Proteins and Their Overall Tertiary Structuresa
|
|
|
|
|
aWithin each functional grouping, proteins known to be homologous are listed on a single line, and proteins that fall within the same tertiary-structure subcategory (for at least one of their domains) are bracketed. In spite of the detailed resemblances commonly found within active sites, the great majority of examples shown no similarity of overall structure.
|
|
The really puzzling fact, however, is that there is one glaring exception to the above pattern: the nucleotide-binding domains, especially the dehydrogenases and kinases. Within that functional group, and within the parallel α/β structures, the distribution is exactly what one would expect from the model in which large groups of proteins share an evolutionary origin (for at least one of their domains). Such a pattern could also be explained by especially stringent selective pressures, although there is no evidence at all that the requirements for nucleotide-binding sites are any more restrictive than for any other function (see Section III,C). Or the pattern could result from a combination of moderate selective pressure and some as-yet-unknown restrictions on folding within this general structure category. Whatever the explanation, it must somehow account for the fact that nucleotide-binding proteins (or, perhaps, enzymes in the glycolytic pathway) appear to be different in some fairly fundamental way from any other functional category sampled so far.
Another general approach that has commonly been taken to the problem of evaluating relationships between proteins is calculation of the minimum root-mean-square difference between superimposed a-carbon positions for the similar parts of the structures. This was initially done by Rossmann () to compare nucleotide-binding domains and other structures (e.g., ). These Cα comparisons in general corroborate and quantitate similarities found by inspection, and sometimes have uncovered relationships no one had previously noticed. Considerable progress has been made recently on ways of evaluating the significance level of Cα superpositions (; ; ). Two logically distinct problems are involved: the first problem is evaluating the significance of a given similarity relative to the probability of its "chance" or "random" occurrence, the second problem is estimating the likelihood that a given significant resemblance was produced by divergent rather than convergent evolution. In practice the two problems are attacked together, because no one is as interested in the more obvious (and highly significant) similarities that all proteins share simply as a result of globularity, covalent bonding, and preferred backbone conformations. Ideally one wants the "control" or reference comparisons to incorporate all nonhistorical constraints that apply to protein structure in general: requirements of overall stability, side chain packing, efficient folding, and all the other factors we do not yet know. Then any closer degree of resemblance can be assumed to be due to an historical evolutionary relationship (with a calculable confidence level).
The apparent objectivity of quantitative comparison methods obscures the fact that we do not yet know nearly enough about either the genetic processes or the stability and folding requirements to be sure the estimated probabilities of relationship are correct within an order of magnitude. Most comparison methods cannot readily allow for insertions and deletions; we know that they are an important factor that should be included, but even if the computational difficulties can be overcome, we simply do not have any idea of the relative likelihood of, for instance, one long versus two short insertions or of whether an insertion that makes a wide spatial excursion is any less likely than one which stays close. Because there are fewer degrees of freedom, spatial equivalence between helices must be less significant per residue than betweenβ strands than between nonrepetitive structure, but we cannot quantitate this effect. Functional resemblance is certainly a strong argument for the significance of a resemblance, but it cannot make a case for divergent rather than convergent evolution. Perhaps the most fundamental difficulty is that it is an a priori assumption, not an empirically determined fact, that closeness of spatial coordinates is a suitable measure of evolutionary distance.
At the same time, we need to know more about the genetic mechanisms that may be influential in protein evolution, since our current paradigms are almost certainly too simple. We need to understand more about the practical consequences of exon-intron organization on the gene and whether it generally correlates with domain divisions or with smaller internal units. It would be useful to know how unusual is the circularly permuted amino acid sequence of favin versus concanavalin A (). And we might consider the possibility, for instance, that the helical portion of the larger domain of hexokinase (see Fig. 108) could "bud off" as an independent protein that would have an historical evolutionary relationship to the doubly wound sheet portion of hexokinase but no structural or sequence resemblance whatsoever. In the worst case, it could be that evaluating probable evolutionary relationships in terms of structural resemblance is not generally possible, because the constraints of stability and folding requirements might turn out to be more demanding than the limitations on rapid evolutionary change. However, one must start out with the more optimistic attitude that a sufficiently varied and open-minded program of structure comparisons will teach us a great deal both about the folding constraints and also about the evolutionary history of proteins.